

President Trump’s nomination of Scott Gottlieb as the next FDA Commissioner has given pharma a reason for calm, though a lot will be on his plate.
FDA
Latest News
Advertisement
Advertisement
Patient advocates are witnessing a shift in the culture of drug development as sponsors and regulatory agencies show more interest in incorporating patient perspectives.
Pressure from the FDA and Congress has resulted in sponsors enrolling more women, elderly and ethnically diverse individuals in clinical studies.
FDA bioresearch monitoring staffs are being prompted to update and refine systems for targeting inspections to key study sites.

The needed resources to implement "Cures" legislation makes it critical that user fee programs for drugs are reauthorized well before they expire on Sept. 30.
The FDA has released a report that documents the value of large, confirmatory Phase III studies in ensuring the safety and efficacy of experimental medical products.
Congress took a major step yesterday towards shoring up FDA operations and biomedical research supported by the National Institutes of Health with House passage of the 21st Century Cures bill.

For those still waiting, the time is now to get compliant on FDA's new data submission requirement.

As the discussion on patient centricity continues, industry personnel are attempting to further define the concept. John Whyte of the FDA sits down with us to explain his perspectives and experiences on the subject.
The FDA approved Sarepta’s Exondys for Duchenne muscular dystrophy despite little evidence of efficacy, leading many to regard the decision as not being a model for future drug development. The challenge now is to see if confirmatory trials show more benefit, or lack of efficacy.

The deadline looms for new FDA requirements that all applications for new drugs and biologics compile and submit clinical trial data electronically. Meanwhile, efforts by the Clinical Data Interchange Standards Consortium (CDISC) to develop consensus-based standards for collecting data are not going unnoticed.
FDA and industry have agreed on a set of recommendations for revising the drug user fee program, which now sits before Congress as U.S. election nears.

The FDA’s objectives regarding pediatric labeling have been misunderstood due to a confusing history on the matter. Data extrapolation can leverage an avenue for providing more comprehensive labeling for pediatric drugs.

Processes for collecting data of serious adverse events (SAEs) and events of special interest (ESI) during clinical trials have been underwhelming for sponsors. Improving these processes using the latest digital advances can result in more complete analyses and more effective decision-making.

Clinical trial noncompliance has always been an industry concern. According to the FDA, the most common compliance deficiencies during inspections include inadequate investigator oversight, protocol deviations, poor record keeping, insufficient investigational product accountability, and issues with subject protection and consenting1.
The need to revise the design, performance and interpretation of clinical research to reflect changing methods and standards is drawing increased attention.

In response to the debate over allowing patients timely access to new therapies and ensuring safety, the EMA launched the PRIority MEdicines (PRIME) scheme in March. The aim of this initiative is to build upon existing regulations in Europe to support product development in cases of unmet medical need.
FDA's breakthrough drug initiative has proven successful to date, but challenges remain in addressing the expectations and patient concerns surrounding candidates in this program.

The need to involve regulators is crucial when the use of electronic data devices impacts the management of patient safety and evaluation of trial endpoints.
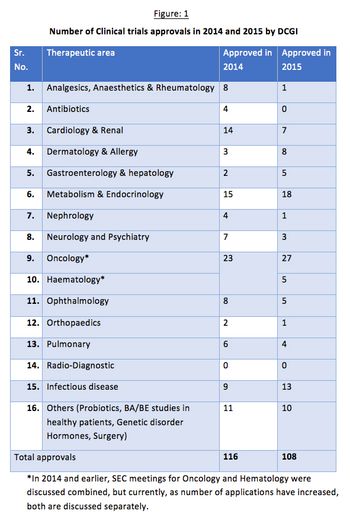
Issues concerning the drug approval process in India have occurred in clinical studies resulting in a reduced interest from sponsors. The Drug Controller General of India (DCGI) has been working to amend and update it’s policies to stimulate growth in this sector.
While thousands of hours are spent in the hopes of finding more effective treatments, many of them are rejected due to administrative errors. These three tips can help reduce the frequency of common administrative mistakes during clinical trials.
The FDA’s breakthrough drug initiative is accelerating clinical development of new therapies. A recent analysis found that pre-market development time for breakthrough-designated drugs is 2.2 years shorter than for those without the designation.
FDA's new commissioner advocates for a "learning healthcare system" and less-complex clinical trial models that tap efficiencies in study design and enrollment.
Agency is asking sponsors to propose demonstration projects that test the use of electronic health records and standards-based technology solutions. Ensuring trials assess new drugs in diverse patient populations is also a priority focus for FDA.

The disruptive influence of data transparency in the status quo of product development may have much longer implications to the healthcare process, and information for patients.
Advertisement
Advertisement
Trending on Applied Clinical Trials Online
1
SCOPE Summit 2026 Keynote Panel: Is Radical Acceleration in Clinical Research Possible?
2
SCOPE Summit 2026 Panel Discussion: Diversity in Clinical Trials—What’s Working, What’s Next
3
Accelerate Clinical Trials with AI-Enhanced Financial Management
4
SCOPE Summit 2026 Keynote Fireside Chat: Aligning Purpose, Innovation, and Operational Excellence in Clinical Development
5