




Ambitious in scale and scope, the Critical Path Initiative provides a rational construct for framing the central paradox of today's new drug and new device development industry.

Ambitious in scale and scope, the Critical Path Initiative provides a rational construct for framing the central paradox of today's new drug and new device development industry.

The Critical Path Initiative (CPI), as it has come to be called, provided the FDA's analysis of the current drug development pipeline problem, characterized by a recent slowdown-instead of the expected acceleration-in innovative medical therapies reaching patients.

In March 2004, FDA issued a provocative white paper: Innovation and Stagnation-Challenge and Opportunity on the Critical Path to New Medical Products. The Critical Path was identified by FDA as those parts of the R&D continuum that constitute bottlenecks in the drug development process.

Scientific discovery is, by its very nature, plagued by a lag between concept development and the acquisition of the tools necessary to fully explore a new technology's application.

When the FDA stated that "the applied sciences needed for medical product development have not kept pace with the tremendous advances in the basic sciences" everyone seemed to agree.

The FDA's Critical Path Initiative acknowledges long-standing and widely accepted challenges facing the clinical research enterprise and identifies several opportunity areas that may help to address these challenges.

Recent events highlight the need to clarify rules and policies for pediatric studies.
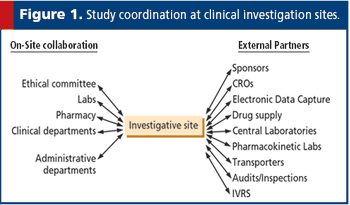
European law now gives prominence to the suitability of investigators and the quality of facilities, two important clinical trial issues.

The country is in the forefront of European Union nations prepared to explore the brave new world of post-CTD research.
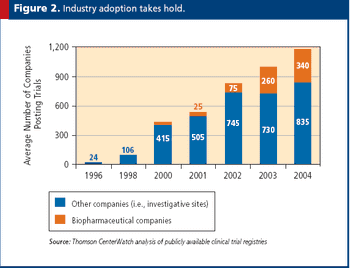
Why it is imperative for government and commercial trial publishers to collaborate in addressing this issue faster.

Years of debate about pediatric research in the EU may finally end up going somewhere.

For truly informed consent, methods should gauge a subject's real grasp of a clinical trial's complexities.
Regulators and researchers seek to curb the overload on IRBs and harmonize AER policies.
For the EU, these new good clinical practice rules come not from Brussels, but Mt. Sinai.

It can be difficult to determine when a hospitalization is "for treatment" and counts as serious under Japanese regulations.
A recent conference featured debate on the role of research ethics committees.
Clinical trials may take longer and become larger as proposals emerge to strengthen regulatory policies.
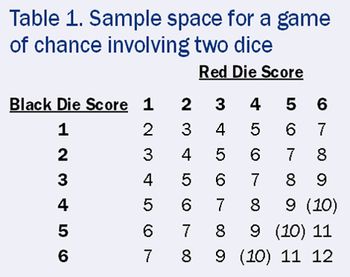
A simple game of chance clears up the confusion over what randomization can and cannot do.

Informed consent takes place in the brain. While that seems obvious, the implications of that statement go much deeper. To a great extent, both bioethics and federal policy are based on 17th century assumptions by philosophers such as Descartes-who knew nothing of the structure and function of the brain.
New policies aim to simplify testing for single-pill combos and products that combine drugs with medical devices.
Concerns over risky medicines are shutting down clinical studies and boosting demand for comparative drug analysis.

Because it can take time to obtain independent EC or review board approval, the temptation to start a study without it can become strong.
Industry?s proposal for a shared database has merit, but is it ?too little too late??

If the activity is a significant risk device study, surgeons must first obtain FDA and IRB approval.

Perhaps a way forward is to refocus our efforts on a "culture of conscience," since our conscience often guides our actions.